Синдром эдвардса
Содержание:
- Эффективность лечения и прогноз
- Причины
- Формы синдрома Эдвардса:
- Синдром Эдвардса – диагностика
- Причины и профилактика синдрома Эдвардса
- Симптомы заболевания
- Лечение синдрома Эдвардса
- Причины развития синдрома Эдвардса
- Типы синдрома Эдвардса: как зависит тяжесть проявлений
- Прогноз
- Причины заболевания
- Проявления синдрома
- Прогноз
- Частота появления
- Вариации
- Проявления синдрома
- Диагностика
- Болезнь Эдвардса на УЗИ
- Методы диагностики синдрома Эдвардса
- Проявления синдрома
Эффективность лечения и прогноз
Специфического метода борьбы с патологией на сегодняшний день нет. Подобная проблема связана с генетической основой заболевания. Поэтому прогноз при синдроме Эдвардса неутешительный. Больше половины детей погибают в первые три месяца после рождения. Лишь малой части, не более 10%, удается дожить до года. Даже если получается спасти пациента, растить его очень сложно. Это связано с высоким риском формирования осложнений, а также выраженной умственной отсталостью, которая с трудом поддается коррекции. Исключение составляют лишь дети с мозаичным кариотипом синдрома Эдвардса. В таких случаях в воспитание хоть и вносятся определенные изменения, зачастую удается добиться высокого качества жизни.
Во многих случаях лечение заболевания предполагает использование хирургических методик. Проводится оперативная коррекция пороков сердца, восстанавливается нормальная работа желудочно-кишечного тракта, ушиваются грыжи. Осуществляется и устранение косметических дефектов, таких как «заячья губа». На этапе лечения детям зачастую требуется зондовое питание, а также внутривенная инфузионная терапия. Поскольку при проведении хирургических вмешательств при синдроме Эдвардса велик риск развития инфекционных заболеваний, оправдано использование антибактериальных средств.
После радикальной коррекции врожденных нарушений потребуется длительная медикаментозная поддержка. Выбор тактики дальнейшего лечения основан на клинических проявлениях заболевания. Используются адреноблокаторы, мочегонные и гормональные препараты. Важную роль в реабилитации играет и правильное питание, которое зачастую возможно только при использовании смесей.
Причины
При отсутствии в анамнезе семьи данного заболевания, при абсолютном здоровьем родителей, всё равно есть риск рождения ребенка с данным заболеванием. Науке известно, что клетка человека включает 46 хромосом. В яйцеклетке и сперматозоиде находится по двадцать три хромосомы. Когда они объединяются, то объединяется и количество хромосом. Причины синдрома Эдвардса на сегодняшний день изучены недостаточно.
Исследователи говорят, что вследствие мутаций генов формируется в 18-й паре хромосом одна лишняя. В 2 случаях данной болезни из 100 восемнадцатая хромосома удлиняется, при этом 47-й хромосомы не формируется, а происходит транслокация.
В трех случаях болезни из ста врачи говорят о мозаичной трисомии. Это значит, что лишняя хромосома присутствует только в части клеток организма плода, а не абсолютно во всех. Но по симптомам три описанные варианта болезни сходятся. Только в первом случае течение может быть тяжелым и больше шансов летального исхода.
Формы синдрома Эдвардса:
• Полная
трисомия 18.
Полная или классическая форма синдрома
Эдвардса предполагает, что все клетки
в организме имеют дополнительную
хромосому. Данный вариант заболевания
встречается более чем в 90% случаев и
является наиболее тяжелым.
• Частичная
трисомия 18.
Частичная трисомия 18 является весьма
редким феноменом (не более 3% от всех
случаев синдрома Эдвардса). При ней в
клетках организма содержится не целая
дополнительная хромосома, а лишь ее
фрагмент. Такой дефект может быть
результатом неправильного деления
генетического материала, но встречается
он очень редко. Иногда часть восемнадцатой
хромосомы присоединяется к другой
молекуле ДНК (внедряется в ее структуру,
удлиняя молекулу, или просто «цепляется»
с помощью мостика). Последующее деление
клеток приводит к тому, что в организме
имеется 2 нормальные хромосомы номер
18 и еще часть генов с этих хромосом
(сохранившийся фрагмент молекулы ДНК).
В этом случае количество врожденных
дефектов будет намного ниже. Наблюдается
избыток не всей генетической информации,
закодированной в 18-й хромосоме, а лишь
ее части. Для пациентов с частичной
трисомией 18 прогноз лучше, чем для детей
с полной формой, но все равно остается
неблагоприятным.
• Мозаичная
форма.
Мозаичная форма синдрома Эдвардса
встречается в 5 – 7% случаев данного
заболевания. Механизм ее появления
отличается от других видов. Дело в том,
что здесь дефект образовался уже после
слияния сперматозоида и яйцеклетки.
Обе гаметы (половые клетки) изначально
имели нормальный кариотип и несли по
одной хромосоме каждого вида. После
слияния сформировалась клетка с
нормальной формулой 46,ХХ или 46,XY. В
процессе деления этой клетки произошел
сбой. При удвоении генетического
материала один из фрагментов получил
дополнительную 18-ю хромосому. Таким
образом, на определенном этапе
сформировался зародыш, часть клеток
которого имеют нормальный кариотип
(например, 46,ХХ), а часть – кариотип
синдрома Эдвардса (47,ХХ, 18+). Доля
патологических клеток никогда не
превышает 50%. Их число зависит от того,
на каком этапе деления начальной клетки
произошел сбой. Чем позже это происходит,
тем меньше будет доля дефектных клеток.
Форма получила название из-за того, что
все клетки организма представляют собой
своеобразную мозаику. Часть из них
здорова, а часть – с тяжелой генетической
патологией. Закономерности в распределении
клеток в организме при этом не наблюдается,
то есть все дефектные клетки не могут
локализоваться только в одном месте,
чтобы их можно было удалить. Общее
состояние пациента при этом легче, чем
при классической форме трисомии 18.
Синдром Эдвардса – диагностика
Описываемая генетическая болезнь является прямым показанием для прерывания беременности. Дети с синдромом Эдвардса никогда не смогут жить полноценно, а их здоровье будет стремительно ухудшаться
По этой причине важно диагностировать трисомию 18 на максимально раннем сроке. Для определения данной патологии разработано несколько информативных скринингов
Анализ на синдром Эдвардса
Существуют неинвазивные и инвазивные методики исследования биологического материала. Второй тип тестов считается самым достоверным и надежным, он помогает выявить синдром Эдвардса у плода на ранних этапах развития. Неинвазивным является стандартный пренатальный скрининг крови матери. К инвазивным способам диагностики относятся:
- Биопсия ворсин хориона. Исследование проводится с 8 недели. Для осуществления анализа отщипывается кусочек оболочки плаценты, потому что его структура почти идеально совпадает с тканью плода.
- Амниоцентез. В ходе тестирования берется образец околоплодных вод. Эта процедура определяет синдром Эдвардса с 14 недели вынашивания.
- Кордоцентез. Для анализа требуется немного пуповинной крови плода, поэтому такой метод диагностики применяется исключительно на поздних сроках, с 20 недели.
Риск синдрома Эдвардса по биохимии
Пренатальный скрининг осуществляется в первом триместре беременности. Будущая мать должна сдать кровь в период с 11 по 13 неделю вынашивания для биохимического анализа. По результатам определения уровня хорионического гонадотропина и плазменного протеина А рассчитывается риск синдрома Эдвардса у плода. Если он высок, женщина вносится в соответствующую группу для следующего этапа исследований (инвазивных).
Синдром Эдвардса – признаки по УЗИ
Указанный вид диагностики используется редко, преимущественно в случаях, когда беременная не прошла предварительный генетический скрининг. Синдром Эдвардса на УЗИ можно выявить только на поздних сроках, когда плод уже почти полностью сформирован. Характерные симптомы трисомии 18:
- внутриутробные пороки сердечно-сосудистой и мочеполовой системы;
- аномалии опорно-двигательных структур;
- патологии костей черепа и мягких тканей головы.
- Косвенные признаки болезни на УЗИ:
- брадикардия;
- задержка развития плода;
- одна артерия в пуповине (должно быть две);
- грыжа в брюшной полости;
- визуальное отсутствие костей носа.
Причины и профилактика синдрома Эдвардса
Синдром Эдвардса возникает из-за миопического нерасхождения хромосом. Процесс осуществляется на стадии сперматогенеза или гаметогенеза. Практически в 90% случаев заболевание представлено простой трипаносомой. Реже возникают несбалансированные перестройки хромосом или мозаичная разновидность заболевания. Причины появления синдрома Эдвардса обычно обусловлены появлением в генетическом коде лишней материнской хромосомы.
Генетики пока не смогли определить, почему возникает подобная хромосомная мутация. Считается, что такое явление случайно, и никакие внешние факторы на него влияние не оказывают. Однако всё же ряд причин, способствующих хромосомным аномалиям, сегодня выявлен. В перечень входят:
- инфекции половых органов у родителей;
- наследственность;
- возраст родителей больше 45 лет;
- в процессе беременности мать и плод были подвергнуты радиоактивному излучению;
- женщина долгое время употреблял медикаменты, оказывающие влияние на иммунную, эндокринную и репродуктивную системы (аналогичное правило действует и в отношении мужчин);
- имеет место быть длительное употребление табака, наркотиков и алкоголя.
Как и все генетические патологии, синдром Эдвардса не поддается предотвращению при помощи профилактических методов. Чтобы снизить риск возникновения заболевания, родители могут только предварительно пройти диагностические исследования, позволяющие заранее определить вероятность появления мутации. Дополнительно во время беременности за женщиной и плодом должен осуществляться строгий контроль. Нельзя пренебрегать процедурой антенатального скрининга и рядом других исследований.
Симптомы заболевания
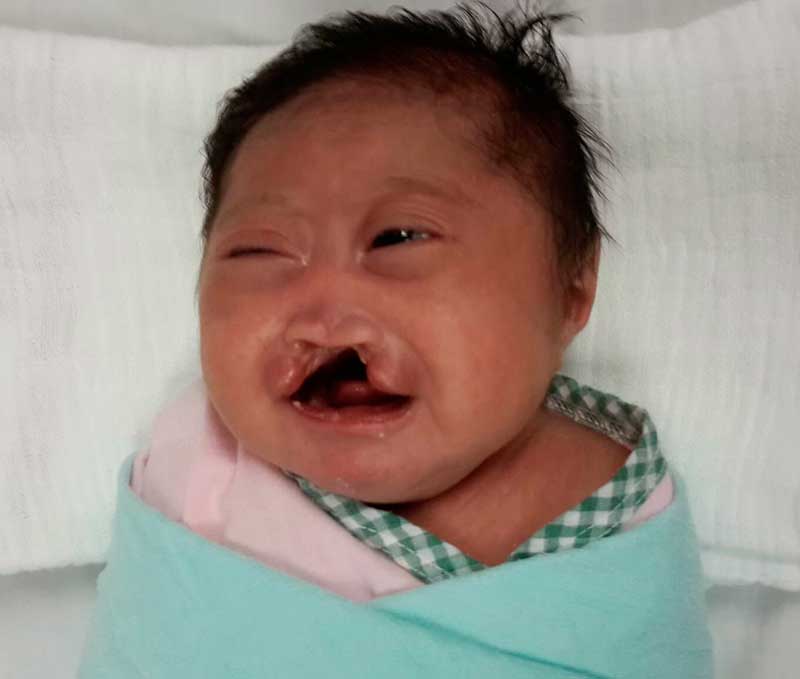
Проявляется синдром у всех детей абсолютно по-разному: у некоторых возникают только основные симптомы заболевания, у других – проявляется их абсолютное большинство.
Различна у пациентов также и степень проявления имеющихся симптомов.
Итак, внешне синдром Эдвардса проявляется следующим образом:
- ребенок имеет маленькую голову по сравнению с туловищем;
- узкие глаза (см. фото выше);
- плохо развитая нижняя челюсть;
- искажение черт и формы лица;
- удлиненные ушные раковины. Часто также отсутствуют некоторые составляющие части уха (мочка, слуховой канал);
- широкая и укороченная грудная клетка;
- верхняя губа совсем небольшого размера, маленький рот;
- сильно расширенная переносица, иногда «вдавленная» внутрь черепа;
- стопы ног неправильной формы;
- высокое небо с разрезом (волчья пасть);
- низкий лоб, затылок сильно выступает;
- косоглазие.
Помимо визуальных признаков, имеются и внутренние, куда более серьезные по своей природе:
- порок сердца;
- наличие грыж;
- отсутствие рефлексов, типичных для маленьких детей;
- недоразвитость мозжечка;
- удвоенные мочеточники;
- почечная недостаточность;
- маленькая масса тела, около 2 кг при рождении;
- с возрастом развивается слабоумие;
- дистрофия мышц;
- онкология различных органов.
Лечение синдрома Эдвардса
Нужно учитывать, что только один из десяти детей, родившихся с подобной патологией, доживают до одного года. Потому первоначальное лечение состоит в избавлении от заболеваний, повышающих риск летального исхода. Если имеет место быть атрезия кишечника или анального прохода, используются меры для дефекации. Ребенка кормят через зонд. Если в организме новорождённого наблюдается активные воспалительные или инфекционные процессы, осуществляется терапевтическое лечение.
Если ребёнка всё же удаётся спасти, в последующем может быть выполнено оперативное вмешательство. В первую очередь выполняется устранение волчьей пасти. Дополнительно проходит борьба с пороками сердца. Пупочная и паховая грыжа удаляются при помощи оперативных методов.
Проводится и медикаментозная терапия. Ребенку назначают лекарства, которые позволяют избавить его от запоров, повышенной кислотности и метеоризма. Родители должны понимать, что синдром Эдвардса способствует появлению следующих патологий:
- отит;
- новообразование в почках;
- гипертензия легких;
- конъюнктивит;
- воспаление лёгких;
- высокое артериальное давление;
- синусит и фронтит;
- инфекционные заболевания мочеполовой системы.
Из-за большого количества патологических изменений, происходящих во внутренних и внешних органах больного, дальнейший прогноз развития неблагоприятен. Те дети, которые сумеют дожить до года и достичь относительно взрослого возраста, будут иметь явное умственное торможение в развитии. Они не смогут самостоятельно ухаживать за собой и удовлетворять свои естественные требования. За такими пациентами необходимо обеспечить постоянный уход и контроль на протяжении всей их жизни.
Однако дети с отклонениями понимают, когда с ними ласково общаются, играют и утешают. Дети с таким синдромом могут научиться самостоятельно есть, улыбаться. Возможно постепенное обучение другим полезным бытовым навыкам.
Высокое количество патологий внутренних органов, развившихся неправильно, приводит к высокому проценту детской смертности. Если в первые месяцы беременности женщина попала в группу риска по этому заболеванию, медики рекомендует выполнить аборт по медицинским показаниям. Однако окончательное решение остается за женщиной.
Причины развития синдрома Эдвардса
Как и две других патологии, указанные выше, синдром Эдвардса относят к генетическим патологиям, при которых обнаруживается дополнительная хромосома в аутосомах (не половые хромосомы). У здорового человека 46 хромосом – 22 пары – аутосомы (неполовые), и одна пара – половые, определяющие – девочка это или мальчик. В хромосомах компактно упакованы нити ДНК, в которой записана вся генетическая информация об организме, начиная с процесса его зачатия и до смерти. Вся программа жизни записана именно в нитях ДНК.
При синдроме Эдвардса организм страдает не от генных дефектов, а хромосомных, возникает дефект в делении половой клетки, которая затем дает ребенку жизнь, и в ней остается лишняя хромосома. При классическом варианте формирования синдрома Эдвардса имеется удвоение 18-ой хромосомы, и родиться могут с подобным синдромом как мальчик, так и девочка. Во всех клетках такого ребенка существует избыток генетической информации. Подобная избыточность ДНК приводит к целому ряду нарушений, которые приводят как к внешним дефектам, так и проблемам в работе систем и органов. Из-за наличия третьей, лишней хромосомы и возникло научное название – трисомия.
Типы синдрома Эдвардса: как зависит тяжесть проявлений
Исходя из того, какого рода хромосомный дефект имеется при синдроме, выделяют три типа течения:
- Полная трисомия по 18-ой паре.
- Частичная трисомия по 18-ой паре
- Мозаичная форма патологии.
При полной форме (она же относится к классическому синдрому) предполагается, что совершенно во всех клетках имеется дополнительная хромосома. Подобный вариант наиболее тяжелый и встречается наиболее часто, прогнозы при нем самые неблагоприятные.
Частичная трисомия относится к редким вариантам патологии, возникает в 3% случаев, при такой проблеме содержится не полная третья хромосома, а только ее кусок. Подобный дефект может образовываться в результате проблем с делением клеток (в особенности их хромосом), что возникает крайне редко. Может быть и вариант того, что лишняя часть 18-ой хромосомы крепится к отдельным молекулам ДНК, приводя к их удлинению и изменению. В таких клетках потом остается две 18-ых хромосомы и часть лишнего материала. Тогда врожденный синдром будет не таким тяжелым, так как в клетках появляется дополнительная информация не всей информации, а только ее части. Прогноз при таком состоянии хотя и неблагоприятный, но течение синдрома гораздо легче, чем при полной форме.
Развитие мозаичной формы проявляется примерно у 5-8% детей с подобной аномалией. Но по механизмам подобный тип синдрома отличается. Интересно, что подобная форма развивается после того, как сперматозоид сливается с яйцеклеткой, и обе получившиеся клетки изначально обладают нормальным набором хромосом – 46ХХ или 46XY. Но при первых же делениях в образованной зиготе образовался сбой и одна из клеток приобрела в силу этого дополнительную хромосому. В результате зародыш приобрел клетки с обычными хромосомами – их 46, другая же часть из них имеет лишнюю хромосому – то есть 47. Количество клеток с лишней хромосомой при таких проблемах не превысит 50%.
Обратите внимание
Чем выше количество дефектных клеток у зародыша и ребенка, тем хуже прогнозы, и зависит их объем от того времени, когда произошел сбой при делении. Чем позже это произошло, тем меньший объем дефектных клеток имеет кроха, и снижено количество проблем развития.
Данная форма получила название мозаичной в силу того, что нормальные клетки перемешаны с дефектными. Атипичные клетки с лишней хромосомой разбросаны хаотично по всему организму, и нет способа, чтобы можно было их каким-либо образом удалить. Эта форма протекает несколько легче, длительность жизни выше.
Прогноз
Абсолютное большинство детей с синдромом Эдвардса гибнет еще в утробе матери из-за того, что организм женщины отторгает плод. В случае, если ребенок родился, прогноз очень неутешителен. Большая часть детей с таким заболеванием живет до нескольких месяцев.
В том случае, если ребенок рождается не с самой серьезной формой заболевания, вероятность прожить до 10 лет резко увеличивается.
Если ребенок прожил уже несколько лет с синдромом Эдвардса, за ним можно заметить полную неактивность и слабо разбитые умственные способности. К сожалению, сделать с этим ничего нельзя, независимо от того, как за ним ухаживают родственники.
Многие родители стараются чему-то обучить своего больного ребенка – это абсолютно бесполезно. Даже тот небольшой процент больных, который достигает довольно зрелого возраста при этой болезни, умеет совсем немного.
В большинстве случаев такие люди могут самостоятельно поднимать голову, узнавать только несколько человек из ближайшего круга (те, кто чаще всего находится рядом), самостоятельно есть, иногда – передвигаться.
Если ребенок или уже взрослый человек начинает ходить, он сильно выгибает ступни внутрь, «косолапит», как говорят.
Стоит отметить, что не для всех детей подходит практика поддержания мышечного тонуса. Некоторым больным это помогает улучшить общее состояние; другим это только навредит ввиду того, что у них могут возникнуть проблемы с опорно-двигательным аппаратом и боли в позвоночнике.
По этой причине необходимо консультировать с лечащим врачом ребенка, чтобы не навредить ему.
Причины заболевания
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления
Частота появления синдрома Эдвардса составляет ~ 1:3000 зачатий и 1:6000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Вариации
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
Проявления синдрома
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Диагностика
Очень большое значение имеет диагностика синдрома Эдвардса, так как данная хромосомная патология — медицинское показание для прерывания беременности. Для выявления заболевания используются следующие методики.
УЗИ и допплерография
Существуют особые признаки синдрома Эдвардса на УЗИ, которые должен заметить врач:
- множественные аномалии развития плода;
- агенезия пупочной артерии;
- малая величина плаценты;
- многоводие.
Но всё это косвенные признаки синдрома Эдвардса при беременности, которые должны быть подтверждены данными других исследований.
Стандартный пренатальный скрининг
Предполагает анализ крови на наличие сывороточных маркеров:
- с 11 по 13 недели: βХГЧ и PAPP;
- с 20 по 24 недели: βХГЧ, свободный эстриола, α-фетопротеин.
Если на основании этих анализов беременная попадает в группу высокого риска, ей предлагается проведение дополнительной диагностики.
Инвазивные методы диагностики
Проводятся в исключительных случаях, когда риск рождения малыша с синдромом Эдвардса очень велик. К инвазивной диагностике относятся:
- биопсия хориона;
- амниоцентез;
- кордоцентез;
- последующее кариотипирование плода.
Если заболевание не было вовремя выявлено, и ребёнок с синдромом Эдвардса всё-таки рождается, он подвергается немедленному всестороннему обследованию.
Диагностика после рождения
Обследование больного ребёнка направлено на выявление пороков развития. Новорождённого с синдромом Эдвардса осматривают разные специалисты:
- неонатолог;
- кардиолог;
- невролог;
- хирург;
- ортопед;
- уролог и др.
Самые важные диагностические исследования, которые выполняются ребёнку с синдромом Эдвардса сразу после его рождения:
- эхокардиография;
- УЗИ почек;
- УЗИ органов брюшной полости.
Какими-то особенностями течение беременности при вынашивании ребёнка с синдромом Эдвардса не отличается. Он так же начинает шевелиться в положенные сроки, бывает даже очень активен, никаких дополнительных токсикозов не наблюдается. Поэтому выявить заболевание в этот период может только современная диагностическая аппаратура. Если же малыш всё-таки родился, вся его недолгая жизнь уйдёт на постоянное лечение сопутствующих пороков.
Болезнь Эдвардса на УЗИ
Всем беременным женщинам предписывается пройти обследование на предмет наличия хромосомных отклонений у плода. Синдром Эдвардса до 12 недель никак не диагностируется, хотя отклонения есть с момент оплодотворения яйцеклетки, но начиная с 12 недели возможно обнаружить характерные для этого заболевания симптомы. К ним относятся:
- Брадикардия (нарушение у плода сердцебиения в виде снижения частоты сердечных сокращений),
- Омфалоцеле (наличие в брюшной полости грыж),
- Отсутствие визуально определенных косточек носа,
- Отсутствие в пуповине одной артерии (в норме в пуповине должно быть две артерии),
- Наличие кисты сосудистых сплетений (киста сама по себе опасности для плода не несет и самопроизвольно исчезает к 26 неделе беременности, но она часто возникает на фоне различных генетических заболеваний у плода, в том числе синдрома Эдвардса).
Наличие хотя бы одного из указанных отклонений выступает поводом для направления беременной женщины на дополнительное исследование. Принудительного исследования женщины не проводится, на все диагностические процедуры и манипуляции она должна давать добровольное согласие
Врач обязан разъяснить смысл и важность диагностических процедур в данном случае, а также порядок их проведения
На более поздних сроках беременности на УЗИ могут быть обнаружены расщепление позвоночника плода вследствие дефекта нервной трубки, увеличение жидкости в воротниковом пространстве, укорочение костей, деформация черепа, изменения в структурах мозга. Эти отклонения свойственным многим хромосомным аномалиям.
УЗИ на выявление хромосомных заболеваний у плода называется скрининговым и проводится врачами, прошедшими специальную подготовку в области генетических аномалий развития. Для этого женщина обычно направляется в иное учреждение для обследования, поскольку не в каждой поликлинике есть необходимое оборудование и специалист с соответствующим уровнем подготовки.
ДополнительноПрограмма скрининга беременных в 1 триместре включает в себя также анализ сыворотки крови, поскольку ни один самый точный прибор УЗИ не может с абсолютной точностью показать наличие или отсутствие заболеваний у плода.
Риск рождения ребенка с хромосомным заболеванием рассчитывается по таким показателям:
- количество ассоциированного с беременностью плазменного протеина А (анализ сыворотки)
- количество свободной β-субъединицы хорионического гонадотропина человека (анализ сыворотки),
- морфологические признаки (УЗИ).
Индивидуальный риск рождения ребенка с генетической аномалией развития 1/100 и выше.
Самый точный диагноз может быть поставлен на основе исследования плодного материала.
Для этого проводится инвазивная процедура. Какая именно процедура будет проведена, зависит от срока беременности:
- В 8-12 недель биопсия ворсин хориона (ткань хориона имеет тот же набор хромосом, что и плод, потому с точностью показывает наличие либо отсутствие генетических отклонений,
- В 14-18 недель амниоцентез (забор и анализ околоплодной жидкости),
- В 18-20 недель и позднее кордоцентоз (забор и анализ пуповинной крови).
Амниоцентез и кордоцентез часто проводятся вместе в ходе одной диагностической процедуры. Это позволяет получить более всесторонние и точные результаты.
Важно В случае постановки диагноза хромосомных нарушений в том числе и синдрома Эдвардса у плода и врожденных пороков развития с неблагоприятным прогнозом для жизни и здоровья ребенка по решению перинатального консилиума и добровольного согласия беременной женщины производится прерывание беременности независимо от срока беременности. Женщина обязательно должна быть проинформирована о диагнозе плода, о течении диагностированного заболевания, особенностях развития и качестве жизни детей, рожденных с подобной аномалией
Женщина обязательно должна быть проинформирована о диагнозе плода, о течении диагностированного заболевания, особенностях развития и качестве жизни детей, рожденных с подобной аномалией.
После 16-18 недель беременности проводится тройной скрининговый тест на хромосомные отклонения плода. В ходе биохимического анализа крови оцениваются такие показатели:
- α-фетопротеин (альфа-ФП);
- свободная β-субъединица хорионического гормона человека (бета-ХГЧ);
- эстриол свободный.
Методы диагностики синдрома Эдвардса
На сегодняшний день существует три типа диагностик синдрома, которые принципиально отличаются друг от друга
Учитывая тот факт, что патология неизлечима и летальна в раннем возрасте, важно обращать внимание на пренатальную диагностику, которая не допускает рождения детей с данными аномалиями. Полезными будут и консультации врачей-генетиков в семьях, где имели место случаи хромосомных патологий. Можно провести:
Можно провести:
- Диагностику половых клеток еще до зачатия. К сожалению, подобный метод применим еще очень ограниченно и только в протоколах ЭКО при предварительном изучении половых клеток. Если же это естественное зачатие, врачи могут прогнозировать более высокие риски рождения таких детей, но это не более, чем прогнозы. Точно узнать – родится ли такой ребенок или нет – невозможно. Исследуется семейный анамнез с учетом всех имеющихся заболеваний, а также оцениваются факторы риска, а также проводится генетический анализ обоих родителей с применением сложной аппаратуры и анализов. За счет определения кариотипа родителей могут делаться определенные выводы.
- Выявление синдрома в период гестации. При развитии плода существует несколько методов прямого или косвенного подтверждения синдрома Эдвардса, изначально для этих целей применяют УЗИ-скрининг и анализы крови с изучением особых показателей, а при высоких рисках проводятся уже инвазивные процедуры – амниоцентез, либо кордоцентез и биопсия ворсинок хориона. Проводят скрининги в строго определенные сроки, когда изменение данных УЗИ и показателей крови будет наиболее значимым.
Важно
Получение непосредственно клеток плода или его крови позволяет провести исследование его хромосом и подтвердить или опровергнуть у него синдром Эдвардса. Это помогает в принятии решения о пролонгации или прерывании беременности по медицинским показаниям.
Подтверждение синдрома после рождения. Его проводят по результатам внешних данных в комбинации с рядом генетических анализов, подтверждающих наличие трисомии по 18-ой паре хромосом
Помимо этого важно оценить состояние ребенка и дать прогнозы в отношении его жизни и состояния здоровья, чтобы понять, какой объем помощи ему требуется для выживания. Без медицинской помощи при подобном синдроме дети могут быстро погибать поле рождения в ближайшие недели
Для выявления пороков во внутренних органах проводят УЗИ и ЭХО-КГ, рентгенографию и ЭЭГ, целый ряд анализов крови и мочи, дополнительные исследования вплоть до МРТ и КТ.
Проявления синдрома
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.




Добавить комментарий