Заболевание вильсона-коновалова
Содержание:
- Обмен меди при болезни Вильсона
- Патологическая анатомия
- Лечение
- Насколько распространена болезнь?
- Клиническая картина и течение
- Этиология и патогенез болезни Вильсона
- Симптомы
- Формы
- Данные статистики
- Формы патологии
- Симптоматика патологии
- Диета
- Возможные осложнения
- Симптомы
- Классификация и стадии развития болезни Вильсона — Коновалова
Обмен меди при болезни Вильсона
Концентрация меди в печени новорожденного в 6—8 раз выше, чем в печени взрослого человека. В первые 6 мес жизни она снижается до 30 мг на 1 г сухой ткани, а затем на протяжении всей жизни остается неизменной благодаря тщательной регуляции всасывания меди в кишечнике, ее транспорта в печень, запасания там с помощью сывороточных и тканевых белков и выведения из организма через желчь.
Всасывание и выведение меди. Среднее суточное потребление меди составляет от 2 до 5 мг, примерно 50% этого количества всасывается в проксимальном отделе тонкой кишки и нековалентно связывается с альбумином плазмы. В печени медь высвобождается и связывается со специфическими «белками, в частности с цитохром-с-оксидазой и церулоплазмином, либо захватывается лизосомами и экскретируется в желчь. Есть два основных пути, посредством которых медь покидает печень.
- Синтез медьсодержащего белка церулоплазмина и его поступление в кровоток.
- Экскреция с желчью.
Генетические нарушения. Повышенное накопление меди при болезни Вильсона обусловлено пониженным выведением ее с желчью, а не с повышенным всасыванием в кишечнике. Причиной являются мутации гена АТР7В, расположенного на 13-й хромосоме. Этот ген кодирует Cu2+-АТФазу, которая экс-лрессируется в печени, почках и плаценте. В результате мутаций нарушается транспорт меди из печени в желчь, и ионы меди накапливаются в гепатоцитах. Cu2+-АТФаза присутствует главным образом в трансцистернах аппарата Голыхжи, где обеспечивает экскрецию меди в желчь, а также ее связывание с церулоплазмином. При недостатке функциональной АТФазы снижается количество меди, которая может связываться с церулоплазмином. В этом случае церулоплазмин, не содержащий меди (апоцерулоплазмин), попав в кровоток, быстро разрушается. Поэтому отличительной чертой болезни Вильсона служит пониженное содержание церулоплазмина в плазме.
Патологическая анатомия
Печень уменьшена и бугриста. Атрофический цирроз печени мало отличается от лаэннековского. Микроскопически участки печени нормального строения, чередуются с некротическими. Отмечаются и регенеративные явления, разрастание рубцовой и молодой соединительной ткани, богатой сосудами. В печени содержится большое количество зёрен меди. Изменения сосудов печени имеют большое значение в нарушении барьерной функции, так как кровь воротной вены может, минуя печень, может поступать прямо в нижнюю полую вену. Наблюдается увеличение селезёнки, гиперплазия её пульпы, явления нефрита, жировое перерождение почечного эпителия, отложение извести.
Изменения в нервной системе расцениваются как ангиотоксические и цитотоксические; поражение мозга не является системным и не ограничивается подкорковыми узлами. Процесс распространяется на различные отделы нервной системы, но изменения в области лентикулярных ядер являются наиболее выраженными и постоянными.
Поражение других отделов имеет следующую последовательность: хвостатое тело, наружный членик бледного шара, глубокие слои мозговой коры, зубчатые ядра мозжечка, ядра гипоталамической области. Остальные отделы мозга поражаются меньше.
Ангиотоксические изменения стоят на первом плане. Они выражаются атонией сосудов, особенно мелких, их расширением, переполнением кровью, образованием стазов, но без развития тромбозов. В различных отделах мозга наблюдаются кровоизлияния. Отмечаются также изменения стенок сосудов с размножением клеток адвентиции и эндотелия, отложением липоидов, гиалинозом капилляров. Вследствие повышенной проницаемости сосудов развивается периваскулярный отёк и аноксическое разрушение нервной ткани. Богатые капиллярами участки мозга поражаются больше всего. Распространение этих изменений в головном мозгу иногда очень обширно, а их характер лучше всего согласуется с представлением о токсическом воздействии крови на сосудистую стенку.
Цитологические изменения выражаются дегенерацией ганглиозных нервных и макроглиальных клеток. Для гепатолентикулярной дегенерации характерно изменение глии (глия Альцгеймера). Глия Альцгеймера образуется из обычных астроцитов, которые бывают двух типов. К первому типу относятся громадные глиозные клетки с бледной протоплазмой и большим, богатым хроматином ядром. Клетки со сморщенным, пикнотическим ядром называют клетками Опальского. Ко второму типу клеток Альцгеймера относятся гигантские ядра, лишенные протоплазмы, которые на окрашенных тионином препаратах представляются «голыми».
Клетки Альцгеймера встречаются в различных отделах мозга, чаще всего в подкорковых узлах, зрительном бугре, гипоталамической области, зубчатом ядре мозжечка. Ганглиозные нервные клетки подвергаются такого же рода изменениям, что и астроциты, которые превращаются в элементы второго типа Альцгеймера.
В печени и извитых канальцах почек обнаруживаются изменения клеток, сходные с альцгеймеровскими. Наблюдаются также нервные клетки с хромолизом в различных стадиях «хронического заболевания» Ниссля (расплавление нервных клеток), реже поражение клеток, определяемых как «тяжёлое» и «отёчное». Воспалительные изменения не выражены. В основе клеточных изменений лежит нарушение клеточного обмена нуклеиновых кислот. Зёрна меди удается обнаружить в различных отделах мозга, больше всего в подкорковых узлах, в наиболее грубо измененных клетках внутри и межклеточно.
Первично страдает печень, изменения в мозгу обусловлены поражением печени. Клинически заболевание печени предшествует неврологическим симптомам.
Лечение
Схема лечения Вильсона-Коновалова подбирается в индивидуальном порядке. Основная задача медикаментозной терапии заключается в выведении избыточного количества меди из организма пациента.
Программа лечения от болезни Вильсона-Коновалова состоит из следующих этапов:
- Диагностика со сбором требуемых анализов и тестов;
- Получение консультации у специалистов с назначением индивидуальной схемы лечения;
- Осуществление медикаментозных и лечебных процедур для устранения симптомов заболевания;
- Прохождение реабилитационных мер в отделении неврологии.
Основные методы лечения Вильсона-Коновалова:
- Прием Д-пенициламина, уменьшающего количество меди в кровяных тельцах больного. Данный вариант лечения относится к патогенетическому;
- Реабилитационные меры в отделении неврологии для устранения негативного воздействия на нервную систему больного;
- Внутренний прием сорбентов и гепатопротекторов (при наличии повреждений печеночных тканей);
- Трансплантация печени при наличии серьезных, не возобновляемых повреждений органа;
- С помощью специализированного оборудования осуществляются процедуры по очистке плазмы крови от токсинов и свободной меди (гемоиммуносорбция), удалению из плазмы продуктов патологического распада (мембранный плазмаферез). Пациенту могут быть назначены до 3 процедур п очистке крови. Данная методика лечения зарекомендовала себя, как максимально эффективная.
Первые результаты от очистки крови заметны уже после 14 календарных дней. Медикаментозное лечение может длиться до полугода.
Кроме того, любая схема лечения от болезни Вильсона-Коновалова сопровождается приемом медикаментов. Среди них чаще всего выписывают:
Лекарственные препараты, снижающие количество меди в организме больного (хелаты);
Медикаменты, способствующие регенерации печеночных клеток;
Желчегонные лекарственные медикаменты;
Комплекс витаминов (важно применение В6);
Лекарства, блокирующие поступление меди в организм с продуктами питания (например, содержащие цинк);
Медикаменты с антиоксидантными свойствами, снижающие интоксикацию организма;
Средства, подавляющие действие иммунной системы;
Лекарственные препараты успокоительного характера для устранения неврологических расстройств;
Средства, обладающие противовоспалительным результатом.
Назначенные доктором медикаменты пациент обязан принимать с начала диагностирования болезни и до конца его жизни. Если больной снизит дозировку или полностью прекратит лечение, то Вильсон-Коновалов может вернуться с более сильной симптоматикой.
В запущенных случаях заболевания, когда медикаментозные методы не дают должного результата, а состояние больного ухудшается диагностирована печеночная недостаточность, назначается трансплантация печени. Новая печень приживается и нормально функционирует уже через месяц после операции.
Насколько распространена болезнь?
Эксперты все еще изучают, насколько распространена болезнь Вильсона. Более старые исследования показали, что больны примерно 1 из 30 000 человек. Эти исследования были проведены до того, как ученные обнаружили мутации генов, вызывающих заболевание.
Более новые исследования генов пациентов предполагают, что заболевание может быть более распространенной. Исследование, проведенное в Великобритании, показало, что примерно у 1 из 7000 человек есть генетические мутации, вызывающие болезнь Вильсона.
Эксперты не уверены, почему генные исследования предполагают, что заболевание встречается чаще, чем считалось ранее. Одной из причин может быть то, что некоторым больным пациентом не ставили диагнозы. Другая причина может заключаться в том, что некоторые пациенты имеют мутации генов, но заболевание по какой-то причине не развивается.
Клиническая картина и течение
Гепато-церебральная дистрофия начинается в детском или молодом возрасте и имеет хроническое прогрессирующее течение. Во многих случаях появлению симптомов поражения нервной системы предшествуют висцеральные расстройства в виде нарушения деятельности печени и желудочно-кишечных расстройств (желтуха, боли в правом подреберье, диспептические явления). Порой развивается выраженный гепато-лиенальный синдром. Со стороны нервной системы на первый план выступают экстрапирамидные симптомы в виде мышечной ригидности, гиперкинезов и расстройств психики. Пирамидные симптомы могут быть, но чаще отсутствуют. Чувствительность обычно не нарушена.
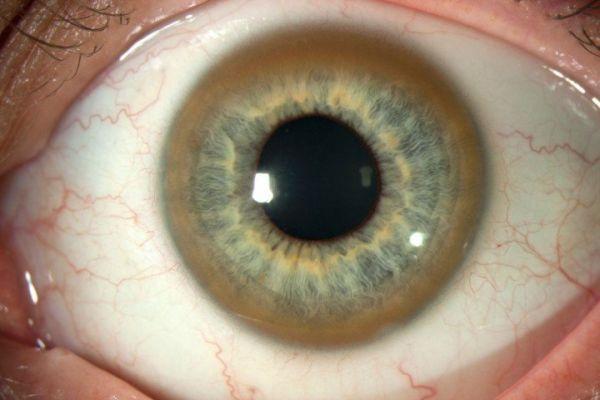
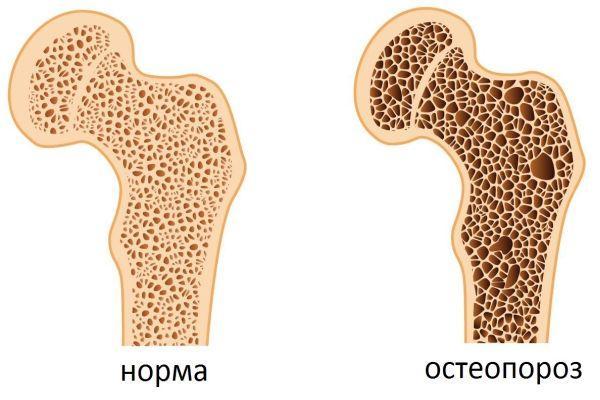
Типичным симптомом болезни являются кольца Кайзера-Флейшера — отложения по периферии роговой оболочки содержащего медь зеленовато-бурого пигмента, более выраженные на поздних стадиях. Иногда отмечается желтовато-коричневая пигментация кожи туловища и лица. Часты геморрагические явления (кровоточивость дёсен, носовые кровотечения, положительная проба жгута), мраморность кожи, акроцианоз. Капилляроскопия обнаруживает атонию капилляров и застойность кровотока. Отмечаются суставные боли, профузные поты, остеопороз, ломкость костей.
Патология печени клинически выявляется примерно у 30 % больных, а в ряде случаев она может быть обнаружена только функциональными пробами, например пробой с нагрузкой галактозой, пробой Квинка, пробой Бергмана-Эльботта, бромсульфофталеиновой пробой; количество билирубина в крови и уробилина в моче обычно увеличено; изменены осадочные реакции Таката-Ара и Грея, обычны лейкопения, тромбоцитопения, гипохромная анемия.
Различают 5 форм гепато-церебральной дистрофии:[уточнить]
- Брюшная форма — тяжёлое поражение печени, приводящее к смерти раньше появления симптомов со стороны нервной системы; заболевают дети. Её продолжительность от нескольких месяцев до 3-5 лет.
- Ригидно-аритмогиперкинетическая, или ранняя форма — отличается быстрым течением; начинается также в детском возрасте. В клинической картине преобладают мышечная ригидность, приводящая к контрактурам, бедность и замедленность движений, хореоатетоидные или торсионные насильственные движения. Характерны дизартрия и дисфагия, судорожный смех и плач, аффективные расстройства и умеренное снижение интеллекта. Заболевание длится 2-3 года, заканчивается летально.
- Дрожательно-ригидная форма встречается чаще других; начинается в юношеском возрасте, течёт медленнее, порой с ремиссиями и внезапными ухудшениями, сопровождающимися субфебрильной температурой; характеризуется одновременным развитием тяжёлой ригидности и дрожания, дрожание очень ритмичное (2-8 дрожаний в секунду), резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает. Иногда обнаруживаются атетоидные хореоформные насильственные движения; наблюдаются также дисфагия и дизартрия. Средняя продолжительность жизни около шести лет.
- Дрожательная форма начинается в возрасте 20-30 лет, течёт довольно медленно(10-15 лет и больше); дрожание резко преобладает, ригидность появляется лишь в конце болезни, а порой наблюдается гипотония мышц; отмечается амимия, медленная монотонная речь, тяжёлые изменения психики, часты аффективные вспышки. Наблюдаются эпилептиформные припадки.
- Экстрапирамидно-корковая форма встречается реже других форм. Типичные для гепато-церебральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжёлым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально.
Наибольшая летальность (50 %) отмечается при печёночной форме с массивным некрозом и гемолизом у детей до 6 лет. Смерть больных от неврологических нарушений при отсутствии лечения наступает через 5-14 лет. Основная причина при этом интеркуррентные заболевания или желудочно-кишечные кровотечения, портальная гипертензия.
Этиология и патогенез болезни Вильсона
Ген, отвечающий за развитие заболевания, расположен в 13 хромосоме. Он участвует в транспортации меди в желчь и включает ее в церулоплазмин. Болезнь Вильсона наследуется, как рецессивный аутосомный признак, и возникает даже при небольшой мутации гена. При таком типе наследования заболеть можно, получив дефектный носитель обязательно от обоих родителей. Люди, имеющие только один пораженный ген, не страдают от болезни Вильсона, но могут ощущать незначительные нарушения метаболизма меди.
В организме здорового человека содержится в среднем не более 100 мг меди, при этом суточная потребность в ней — 1-2 мг. Излишняя часть вещества абсорбируется печенью и выводится с желчью. Болезнь Вильсона приводит к нарушению сразу двух процессов: биосинтеза церулоплазмина — белка, связывающего медь, и ее выведения естественным путем. Из-за этого допустимая концентрация вещества в организма значительно повышается, происходит его отложение в различных органах:
- почках;
- роговице глаза;
- печени;
- головном мозге.
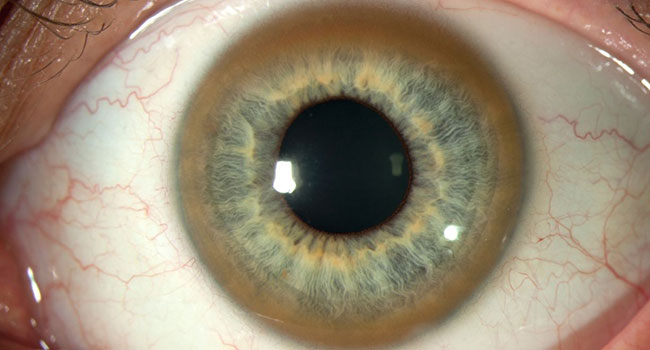 Роговица глаза
Роговица глаза
Симптомы
В организме больного фиксируется повышенное количество меди, что негативно сказывается на работе печени, головного мозга, нервной системы, сердца. Пациент ощущает аритмию (повышенное сердцебиение), развивается цирроз печени, наблюдаются постоянные спазмы и периодическое нарушение речи. Также больной замечает снижение уровня интеллекта и короткие провалы в памяти.
Среди первых видимых симптомов наличия Вильсона-Коновалова выделяют:
- «Кольцо» зелено-бурого цвета на радужке глаза. Такая окантовка образуется из-за чрезмерного содержания меди в человеческом организме. Кроме того, это может быть первый признак отравления медью. Иногда возникает у людей, употребляющих пищу из медной посуды;
- Кожные покровы начинают желтеть, затем желтушность может быть обнаружены на слизистых и на «глазных яблоках»;
- Расстройство желудка, как следствие интоксикации организма. Печень уже не функционирует в нормальном режиме и не способна справиться с объемом меди;
- Движения конечностями в повторяющемся режиме;
- Болевые ощущения справа под ребрами, увеличение печени;
- Температура тела в пределах 37-37,5°С в течение нескольких недель. Также, возможны резкие скачки и перепады температуры до 38-41°С;
- Кожные покровы становятся сухими и шелушащимися;
- Частичный или полный паралич конечностей (рук или ног);
- Припадки судорожного характера. Нередки обморочные состояния;
- Наблюдается агрессивное поведение, расстройство сна и депрессивные наклонности;
- Периодическое возникновение судорожного состояния конечностей;
- Потеря памяти;
- Проблемы с речью, координацией и управлением телом.
Формы
В 1921 году доктором Галлем было описано заболевание клинически похожее на рассеянный склероз. Но, кроме нарушений в работе ЦНС, наблюдалась яркая дегенерация печени. Заболевание получило название гепатолентикулярная дегенерация. Но, с последующими исследованиями доктора Коновалова, было обнаружено, что изменения мозга во время течения заболевания не ограничивались лентикулярной дегенерацией. В 1960 году болезнь получила новое название – «гепатоцеребральная дистрофия», однако в некоторых справочниках всё ещё встречается и прежнее её имя.
Болезнь Вильсона это заболевание, характеризующееся нарушением обмена меди, а именно её накоплением в организме. Единого течения она не имеет, поэтому разработаны критерии, определяющие форму течения болезни. Это степень проявления, особенности симптоматики и возраст начала заболевания. Варьированием этих критериев выделяют пять форм.
Брюшная форма – по проявлению самая ранняя форма. Встречается у детей от нескольких месяцев и до пяти лет. Эта форма с очень быстрым течением и всегда летальным исходом от тяжелого поражения печени. Гепатоцеребральная дистрофия происходит настолько быстро, что неврологические нарушения даже не успевают проявиться. Болезнь Вильсона в этой форме лечению не подлежит.
Ригидно-аритмогиперкинетическая форма – ране выявляемая и быстропротекающая форма. Основные симптомы проявляются не в нарушениях работы печени, а расстройстве нервной системе. Типичными симптомами для ригидно-аритмогиперкинетической формы отмечают контрактуры, замедленность движений, дисфагию, судорожный плач, аффективные расстройства. Обычно ребенок не доживает до возраста трех лет.
Дрожательно-ригидная форма – наиболее часто встречаемая форма, обнаруживающаяся в юношеском возрасте. В отличие от вышеописанных подвидов болезни, дрожательно-ригидная форма развивается гораздо медленнее, поддается лечению и корректировке. У этой формы весьма характерный способ протекания с длительными ремиссиями и неожиданными ухудшениями состояния больного. По симптоматике наблюдается гепатолентикулярная дегенерация, дисфагия, дизартрия. С началом первых проявлений заболевания человек может прожить ещё около шести лет.
Признаки дрожательной формы обнаруживаются обычно во взрослом возрасте. После двадцати лет у больных отмечается амимия, тяжелые нарушения психики, возможны эпилептические припадки. Ригидность наблюдается лишь в конце болезни. Эта форма также развивается крайне медленно – в течение 10-15 лет. При своевременном медицинском вмешательстве и пожизненной терапии проявления этой формы заболевания могут полностью сглаживаться.
Экстрапирамидно-корковая форма – редкое проявление патологии. Болезнь Вильсона в такой форме представляет собой гепатоцеребральную дистрофию с развивающимися пирамидальными парезами, слабоумием и эпилептическими припадками. Больной редко проживает больше восьми лет.
Данные статистики
Последнее время диагностируют больше случаев заболевания. 0,56% населения Земли являются носителями патологически измененного гена.
В тех регионах, где практикуют браки между близкими родственниками, частота заболевания выше. Заболеваемость не зависит от половой принадлежности. Но у детей и молодежи оно диагностируется чаще. Симптомы могут проявиться к 19-20 годам. До пяти лет симптомы могут полностью отсутствовать.
Если патологию не лечить, летальность составляет 100%. Такие больные редко доживают до 30-ти. Причина летальности – геморрагические осложнения, печеночная, почечная недостаточность.
Формы патологии
Существует несколько форм гепатоцеребральнойдистрофии, которые проявляются в соответствии с особенностями ее развития.
Они могут быть:
- Дрожательной. Развитие патологического состояния диагностируется в возрасте 10-30 лет. Заболевание сопровождается тремором. При патологии отмечают развитие брадикинезии и брадилалии. Болезнь сопровождается тяжелым психоорганическим синдромом и эпилептическими приступами.
- Брюшной. Эта форма заболевания появляется у людей в возрасте до 40 лет. Патологический процесс тяжело поражает печень. Это проявляется симптоматикой таких заболеваний, как фульминантный гепатит, цирроз печени, хронический гепатит.
- Ригидно-аритмогиперкинетической. Диагностируется заболевание наиболее часто у детей. На начальном этапе патологический процесс сопровождается амимией, мышечной ригидности. У ребенка становится смазанной речь. Ему сложно выполнять задачи, связанные с мелкой моторикой. В период протекания заболевания умеренно снижается интеллект. Эта форма болезни обладает прогрессирующим течением. Она характеризуется наличием периодов обострений и ремиссий.
- Экстрапирамидно-корковой. Является достаточно редким заболеванием, которое сопровождается эпилептическими припадками. При патологии диагностируют развитие пирамидных и экстрапирамидных нарушений. В период протекания болезни наблюдают появление выраженного интеллектуального дефицита.
Существует несколько форм патологического процесса, которые рекомендуется определять для разработки действенной схемы лечения.
Симптоматика патологии
Болезнь Вильсона наиболее часто диагностируется у пациентов, возрастом 10-25 лет. Патологичесмкий процесс сопровождается:
- мышечной слабостью;
- дрожью;
- деменцией.
У пациентов отмечается различная степень выраженности слабости мышц, которая зависит от степени тяжести патологии. В некоторых случаях возникновение характерного парксонического синдрома наблюдается во всей системе мускулатуры. По своему внешнему виду лицо пациента напоминает маску. У больных диагностируется отвисание нижней челюсти, что приводит к невнятности речи.
При патологическом процессе отмечается нарушение процесса глотания, что объясняется расслаблением мышц носоглотки. Болезнь Вильсона сопровождается усилением слюноотделения. В период протекания заболевания появляется заторможенность двигательной активности. Верхние и нижние конечности замирают в неестественной позе. Слабость мышц приводит к возникновению характерной дрожи.
Слабость мышц приводит к возникновению заметной дрожи, особенно в руках. Если человек отводит руки в стороны или поднимает их на уровень плеч, то это приводит к появлению махов. Возникновение дрожи диагностируется в пальцах в одной или двух верхних конечностях одновременно. На поздних стадиях протекания заболевания диагностируется появление эпилептических приступов. Некоторые больные впадают в кому. При патологии значительно снижаются умственные способности и нарушается психика.
В период протекания заболевания не наблюдается нарушений со стороны чувствительности кожных покровов. Сухожильные рефлексы остаются в норме или повышаются. В некоторых случаях отмечается появление рефлекса Бабинского, который проявляется патологическим разгибанием большого пальца нижней конечности, если умышленно раздражается стопа.
Частым симптомом заболевания является роговичное кольцо. Оно проявляется в виде пигментированной линии, которая имеет коричнево-зеленоватый оттенок. Местом ее расположения является задняя часть роговицы, которая граничит со склерой. Ее заметно офтальмологу при обычном осмотре. При слабой выраженности симптома для его определения используется щелевой осветительный прибор. Заболевание может сопровождаться тромбоцитопенией, анемией и другими изменениями в составе крови, что объясняется нарушением в работоспособности печени.
Симптоматика патологического процесса является ярко выраженной, что предоставляет возможность его самостоятельного определения. Несмотря на это, при появлении первых признаков патологии больной должен обратиться к доктору, который после проведения соответствующих обследований назначит действенное лечение.
Диета
Люди с болезнью Вильсона должны придерживаться определенной диеты, дабы не нагружать печени и не провоцировать обострение недуга.
В рацион необходимо включить:
- Много быстро и легкоусвояемых белков — например, молоко, рис.
- Необходимое количество углеводов (не должно превышать физиологическую норму) — от 250 до 580 грамм в сутки.
- Продукты с содержанием пищевых волокон — макароны, хлеб, неочищенный рис, фрукты, овощи.
- Сложные углеводы. Овощи и зелень, богатые клетчаткой, крахмалом, пектином. Например, морковь, свекла, огурцы, сельдерей, картофель, тыква, капуста и т.д.
- Фрукты и ягоды.
Необходимо снизить суточное потребление жиров. Правильное приготовление пищи, также будет способствовать улучшению состояния больного. Нужно кушать измельченную либо до готовки, либо после пищу. Можно варить, запекать, готовить на пару, но ни в коем случае не жарить.
Естественно, ни одна диета не обходится без запрещенных продуктов, и диета при БВ — не исключение.
В этой ситуации нужно исключить все продукты, содержащие медь:
- все бобовые;
- ракообразных, морепродукты;
- минеральную и любую газированную воду;
- кофе;
- курицу;
- утку, гуся;
- баранину, мясо свиньи;
- колбасу;
- печень;
- грибы;
- перец болгарский;
- щавель;
- орехи, сухофрукты;
- мёд;
- какао, шоколад.
При раннем начале терапии прогноз очень благоприятен. Главное не бояться и терпеливо соблюдать все назначения врача.
Возможные осложнения
В связи с тем, что болезнь поражает печень, нервную систему, вероятные осложнения делятся на такие основные группы:
- Тяжелые заболевания печени. К одним из них относят цирроз, возникающий у большинства пациентов. Прогрессирует он медленно, сопровождается желтушностью кожных покровов, деформацией пальцев рук и ног, расширенными венами на передней брюшной стенке, отеками голеней. Часто больные страдают от кровотечений, возникающих в желудке. Развивается печеночная недостаточность, симптомами которой являются сонливость, поведенческие расстройства, на последней стадии — кома.
- Смерть. Летальный исход ожидает более 70% больных, страдающих от печеночной недостаточности, в особенности — фульминантной.
- Неврологические нарушения. Сюда относят мышечную дистонию, дизартрию, расстройства личности и поведения, эпилептические припадки.
- Невозможность забеременеть у женщин.
Симптомы
Клинические проявления коррелируют с формой заболевания. Для печеной формы типичны признаки: желтуха (желтушное окрашивание кожных покровов и видимых слизистых оболочек, что обусловлено повышением концентрации билирубина в крови), асцит (брюшная водянка, избыточное скопление свободной жидкости в брюшной полости), отеки мягких тканей.
Другие проявления включают геморрагический синдром (кожная геморрагия, проявляющаяся кровоподтеками на коже, кровоточивость слизистых оболочек, что обусловлено нарушением процесса гемостаза), пальмарную эритему (гиперемия кожных покровов ладоней), аменорею (отсутствие менструации у женщин), гинекомастию (доброкачественное увеличение грудных желез у мужчин). Для печеночной формы характерно бессимптомное длительное течение.
Болезнь дебютирует печеночной недостаточностью фульминантного типа, для которой типично молниеносное развитие. При болезни Вильсона-Коновалова поражаются подкорковые отделы ЦНС, что предполагает наличие симптомов, характерных для сферы неврологии. Неврологическая форма сопровождается экстрапирамидными и мозжечковыми расстройствами.
Болезнь проявляется гиперкинезами (патологические неконтролируемые движения, обусловленные ошибочными командами головного мозга), тремором (дрожанием), затрагивающим проксимальные (расположенные ближе к центру тела) отделы конечностей и голову, ригидностью (твердостью и неподатливостью) мышц по пластическому типу. Другие симптомы:
- Гипертонус экстензоров (разгибателей) ног, что приводит к нарушению походки.
- Гипомимия (отсутствие или ослабление лицевой мимики).
- Гипокинезия (ограничение произвольной двигательной активности, уменьшение темпа и объема произвольных движений).
При неврологической форме выявляются такие признаки, как контрактура (ограничение подвижности в суставах, при БВК чаще в области конечностей), дисфагия (нарушение функции глотания), дизартрия (нарушение речи), эпилептические приступы. У 20% пациентов выявляются психические расстройства, которые чаще выражаются в форме депрессии или психоза. Нередко заболевание дебютирует с признаков психических нарушений.
При смешанной форме болезни Вильсона-Коновалова наблюдаются признаки дисфункции печени в совокупности с неврологической симптоматикой – исследование в формате МРТ показывает характерные структурные изменения в мозговом веществе и тканях печени. Заболевание часто сопровождается синдромом холестаза (сокращение объема поступающей в двенадцатиперстную кишку желчи, что обусловлено нарушением процесса ее продукции или экскреции), спровоцированным патологическими процессами в печени.
Для синдрома холестаза характерны признаки: желтуха, кожный зуд, ахоличный стул (кал светлого цвета), нарушение липидного обмена, уменьшение массы тела. Параллельно выявляется гиповитаминоз (дефицит витаминов) A (ослабление, утрата способности видеть в темноте), D (остеопороз, подверженность переломам, нарушение осанки), E (мышечная слабость, нарушения в работе ЦНС), K (геморрагический синдром – склонность к появлению кровоподтеков на коже, кровоточивость слизистых оболочек), печеночная недостаточность.
У 10% пациентов клиническая картина дополнятся признаками почечной недостаточности, что проявляется в форме гематурии (присутствие крови в моче) и гликозурии (наличие глюкозы в моче). У 10-15% больных наблюдается гемолитическая анемия (преждевременный распад эритроцитов, провоцирующий дефицит красных кровяных клеток, переносящих кислород). В 20% случаев у пациентов выявляется остеопороз (уменьшение плотности костной ткани).
Заболевание может протекать на фоне гепатомегалии (патологическое увеличение размеров печени). В некоторых случаях сохраняются нормальные геометрические параметры печени. Болезнь протекает в несколько этапов. В латентный (скрытый) период, который длится 5-7 лет, симптоматика отсутствует.
Классификация и стадии развития болезни Вильсона — Коновалова
В России чаще всего применяется классификация, которая построена на клинических особенностях болезни, сочетания поражения печени и центральной нервной системы. Течение болезни Вилсона — Коновалова подразделяют на:
- бессимптомную форму;
- печеночную форму;
- церебральную форму;
- смешанную форму.
Также применяется классификация Коновалова, которая включает пять форм гепато-церебральной дистрофии:
- Брюшная (абдоминальная) форма — тяжёлое поражение печени, которое проявляется гепатопатией, вильсоновским гепатитом, циррозом печени и фульминарной печёночной несостоятельностью. Может привести к смерти до появления симптомов со стороны нервной системы. Продолжительность от нескольких месяцев до 3-5 лет;.
- Ригидно-аритмогиперкинетическая (ранняя) форма — характеризуется быстрым течением и начинается в детском возрасте. Среди симптомов преобладает мышечная скованность, приводящая к изменениям суставов и их тугоподвижности. Движения замедляются, руки и ноги могут непроизвольно двигаться спирально и червеобразно в сочетании с быстрыми непроизвольными сокращениями мышц. Характерны нарушения речи (дизартрия) и глотания (дисфагия), насильственный, непроизвольный смех и плач, нарушения эмоционального состояния, умеренное снижение интеллекта. Заболевание продолжается 2-3 года, заканчивается смертельным исходом.
- Дрожательно-ригидная форма встречается чаще остальных; начинается в юношеском возрасте, протекает медленно, иногда с периодами полного или неполного восстановления и внезапными ухудшениями, сопровождающиеся повышением температуры тела до 37–38 °C; характерно одновременное развитие тяжёлой скованности мышц и ритмичного дрожания частотой 2-8 подёргиваний в секунду. Эти симптомы резко усиливаются при движениях и волнении, но исчезают в покое и во сне. Иногда наблюдаются дисфагия и дизартрия. Средняя продолжительность жизни около 6 лет.
- Дрожательная форма проявляется с возраста 20-30 лет, течёт относительно медленно (10-15 лет и более); преобладает дрожание, ригидность появляется лишь в конце болезни, иногда наблюдается пониженный тонус мышц; отмечается отсутствие мимики, медленная монотонная речь, тяжёлые изменения психики, частые эмоциональные вспышки, судорожные припадки.
- Экстрапирамидно-корковая форма встречается реже других. Типичные для гепато-церебральной дистрофии нарушения в дальнейшем осложняются внезапно развивающимися двигательными расстройствами по типу параличей (пирамидными парезами), судорожными (эпилептиформными) припадками и слабоумием тяжёлой степени. Длится 6-8 лет, заканчивается летально.
По течению заболевание можно разделить на две сменяющиеся стадии:
- латентная — характеризуется отсутствием внешних проявлений болезни, характерные изменения определяются только при лабораторном исследовании;
- стадия клинических проявлений — появляются специфические симптомы болезни гепато-церебеллярной дегенерации.
Во время лечения выделяют также стадию отрицательного баланса меди, при которой наблюдается регресс клинической симптоматики и характерных лабораторных изменений .








Добавить комментарий